
Objetivos
Describir las técnicas empleadas en el diagnóstico genético y sus limitaciones
Metodología
Descripción de un caso y revisión bibliográfica
Resultados
Mujer de 26 años primigesta sin antecedentes personales de interés. Cribado primer trimestre de bajo riesgo. Serología negativa. Se deriva a la Unidad de Diagnóstico Prenatal por sospecha de displasia esquelética en la ecografía morfológica de 20 semanas.
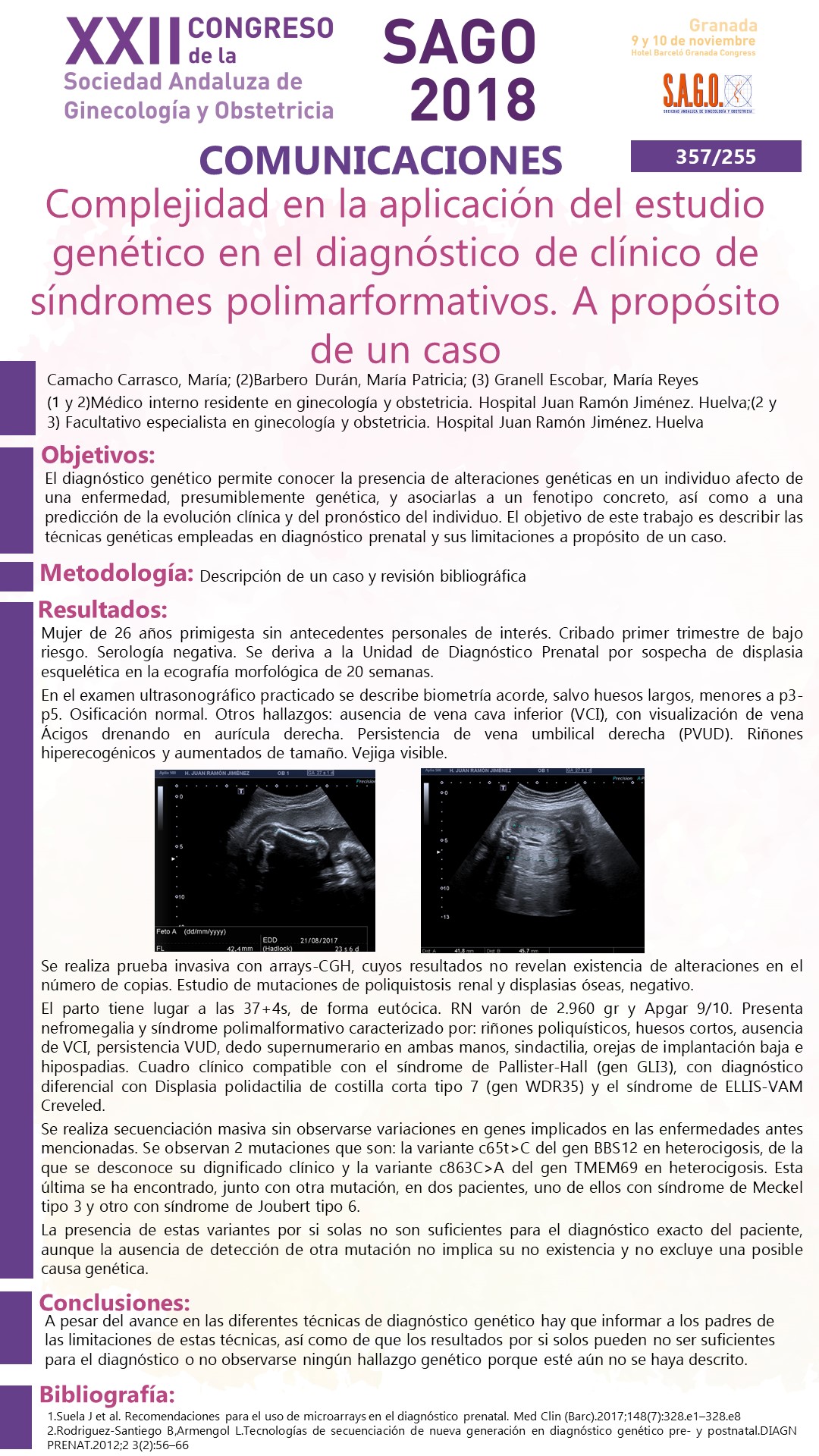
En el examen ultrasonográfico practicado se describe biometría acorde, salvo huesos largos, menores a p3-p5. Osificación normal. Otros hallazgos: ausencia de vena cava inferior (VCI), con visualización de vena Ácigos drenando en aurícula derecha. Persistencia de vena umbilical derecha (PVUD). Riñones hiperecogénicos y aumentados de tamaño. Vejiga visible.
Se realiza prueba invasiva con arrays-CGH, cuyos resultados no revelan existencia de alteraciones en el número de copias. Estudio de mutaciones de poliquistosis renal y displasias óseas, negativo.
El parto tiene lugar a las 37+4s, de forma eutócica. RN varón de 2.960 gr y Apgar 9/10. Presenta nefromegalia y síndrome polimalformativo caracterizado por: riñones poliquísticos, huesos cortos, ausencia de VCI, persistencia VUD, dedo supernumerario en ambas manos, sindactilia, orejas de implantación baja e hipospadias. Cuadro clínico compatible con el síndrome de Pallister-Hall (gen GLI3), con diagnóstico diferencial con Displasia polidactilia de costilla corta tipo 7 (gen WDR35) y el síndrome de ELLIS-VAM Creveled.
Se realiza secuenciación masiva sin observarse variaciones en genes implicados en las enfermedades antes mencionadas. Se observan 2 mutaciones que son: la variante c65t>C del gen BBS12 en heterocigosis, de la que se desconoce su dignificado clínico y la variante c863C>A del gen TMEM69 en heterocigosis. Esta última se ha encontrado, junto con otra mutación, en dos pacientes, uno de ellos con síndrome de Meckel tipo 3 y otro son sd de Joubert tipo 6.
Conclusiones
La presencia de estas variantes por si solas no son suficientes para el diagnóstico exacto del paciente, aunque la ausencia de detección de otra mutación no implica su no existencia y no excluye una posible causa genética.
Bibliografía
1.Suela J et al. Recomendaciones para el uso de microarrays en el diagnóstico prenatal.Med Clin (Barc).2017;148(7):328.e1�–328.e8
2.Rodriguez-Santiego B,Armengol L.Tecnologías de secuenciación de nueva generación en diagnóstico genético pre- y postnatal.DIAGN PRENAT.2012;2 3(2):56�–66
María Camacho Carrasco
Médico Residente de Obstetricia y Ginecología. Hospital Juan Ramón Jiménez. Huelva.
María Patricia Barbero Durán
FEA. Obstetricia y Ginecología. Hospital Juan Ramón Jiménez. Huelva.
María Reyes Granell Escobar
FEA. Obstetricia y Ginecología. Hospital Juan Ramón Jiménez. Huelva.
Todos los derechos Reservados © 2022
